Содержание
- Сопровождение государственной регистрации медицинских изделий в России
- Комплексное сопровождение государственной регистрации российских медизделий включает:
- Для медицинских изделий зарубежного производства дополнительно выполняется:
- Стоимость комплексного сопровождения регистрации мед. изделий от 320 000 руб.
- Для выставления коммерческого предложения просим указать следующую информацию:
- СПРАВОЧНАЯ ИНФОРМАЦИЯ:
- Государственная регистрация медицинских изделий
- Нормативно-правовое основание регистрации
- Сроки регистрации
- Документы, необходимые для регистрации медицинского продукта
- Как проходит разработка документации в ООО «Sigma Lab»?
- Сотрудничество с ООО «Sigma Lab»
- Плюсы сотрудничества с компанией «Sigma Lab»
- Как проверить легальность и регистрацию лекарства, БАДа?
- Как не купить пустышку и не легальный лекарственный препарат?
- Как проверить лекарство через Минздрав России?
- Как проверить лекарство на сайте Росздравнадзора?
- Как проверить БАДы на подлинность на сайте Роспотребнадзора?
- Как проверить лекарство и БАД через справочник лекарств РЛС?
- Правительство утвердило перечень медицинских изделий для упрощенной госрегистрации
Сопровождение государственной регистрации медицинских изделий в России
В нашей компании Вы можете заказать комплексное сопровождение процесса регистрации медицинских изделий, а также выполнение отдельных этапов данной процедуры.
Комплексное сопровождение государственной регистрации российских медизделий включает:
- Первичную оценку документов и внесение исправлений, при необходимости;
- Подготовку и организацию токсикологических испытаний;
- Подготовку и организацию технических испытаний;
- Подготовку и подачу регистрационного досье в Росздравнадзор;
- Проработку замечаний от экспертной организации;
- Подготовку к проведению клинических испытаний;
- Возобновление процесса регистрации (подача результатов клинических испытаний);
- Получение регистрационного удостоверения Росздравнадзора.
Для медицинских изделий зарубежного производства дополнительно выполняется:
- Получение разрешения на ввоз образцов;
- Организация и проверка перевода документации на медизделие.
Стоимость комплексного сопровождения регистрации мед. изделий от 320 000 руб.
Примечание: Перевод, доработка документации, нотариальное заверение, госпошлина, экспертиза безопасности и качества, технические, токсикологические и клинические испытания в стоимость не включены и оплачиваются отдельно.
Окончательная стоимость определяется после получения информации о медицинском изделии, которое требуется зарегистрировать и проработки перечня услуги, необходимых для успешного прохождения данной процедуры.
Для выставления коммерческого предложения просим указать следующую информацию:
- Название МИ;
- Его назначение;
- Область применения;
- Принцип действия МИ;
- Эксплуатационная документация (если есть);
- Зарегистрированные аналоги (если есть);
- Варианты исполнения (если есть).
СПРАВОЧНАЯ ИНФОРМАЦИЯ:
Государственная регистрация медицинских изделий в РФ осуществляется регистрирующим органом — Федеральной службой по надзору в сфере здравоохранения (Росздравнадзор) на основании оценок, подтверждающих качество, эффективность и безопасность изделий.
После успешного прохождения процедуры государственной регистрации, на медицинское изделие выдается регистрационное удостоверение Росздравнадзора и сведения о нем вносятся в Государственный реестр изделий медицинского назначения и медицинской техники. Удостоверение может быть выдано как на отечественную, так и на зарубежную компанию, зарегистрированную, согласно требованиям Российского законодательства.
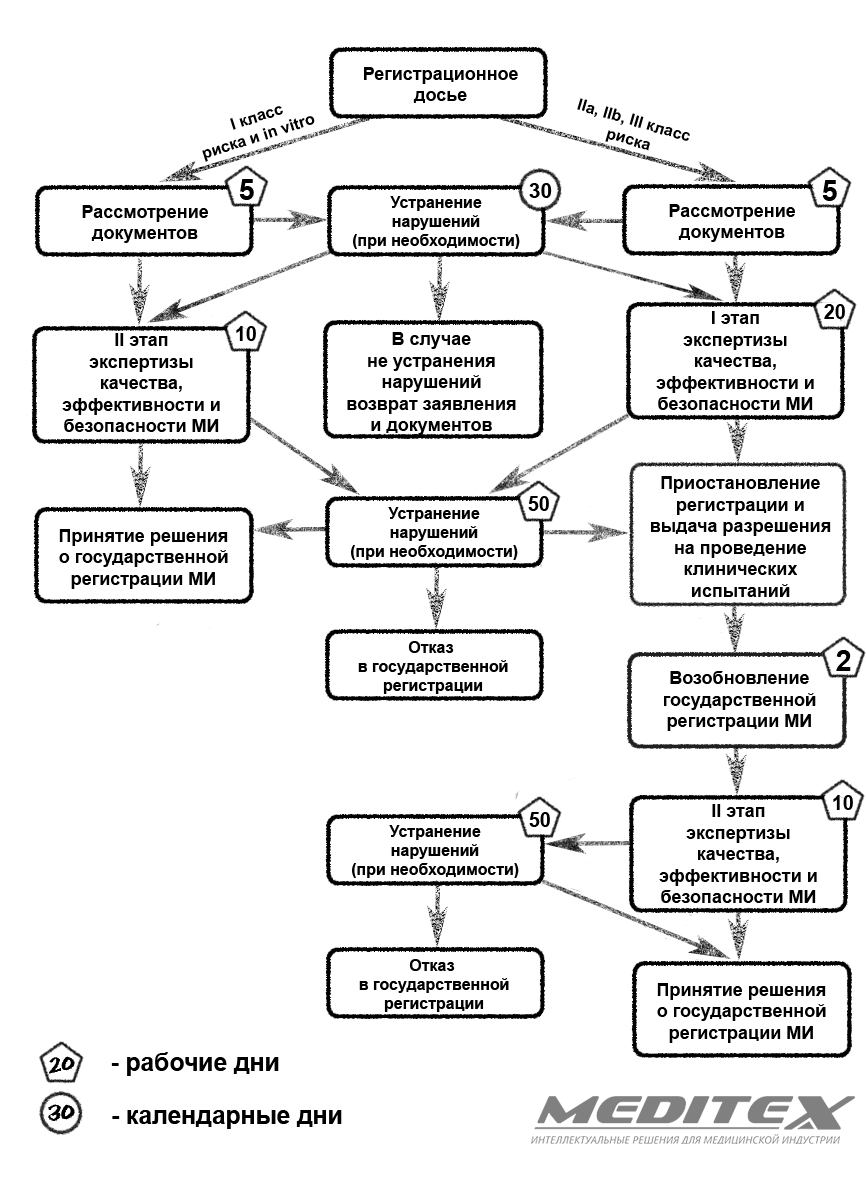
Этапы государственной регистрации медицинских изделий

1. Подготовка документации
1.1. Первичная оценка документации на соответствие требованиям Росздравнадзора.
1.2. Корректировка документации в соответствии требованиям Росздравнадзора.
1.3. Получение разрешения на ввоз образцов (только для изделий зарубежного производства)
1.4. Испытания
1.4.1. Проведение токсикологических испытаний
1.4.2. Проведение технических испытаний.
1.5. Формирование и подача досье в Росздравнадзор.
2. Экспертиза в Росздравнадзоре (I этап)
2.1. Комплект регистрационной документации в Росздравнадзоре проходит проверку полноты и достоверности предоставленных сведений.
2.2. Экспертиза качества, эффективности и безопасности проводится экспертной организацией в срок не превышающий 20 рабочих дней.
2.3. По окончании экспертизы составляется заключение о возможности (невозможности) проведения клинических испытаний (для медицинских изделий класса риска 2а, 2б и 3).
3. Подтверждение клинических данных
3.1. Проведение клинических испытаний
3.2. Подача клинических данных
4. Экспертиза в Росздравнадзоре (II этап)
4.1. Экспертиза клинических данных в срок не превышающий 10 рабочих дней
4.2. По окончании экспертизы принимается решение о выдачи регистрационного удостоверения
Примечание: I и II этап экспертизы для медицинских изделий 1 класса риска и in vitro объединены
Классы риска изделий
В соответствии с приказом Министерства здравоохранения от 6 июня 2012 г. N 4н медицинские изделия в зависимости от потенциального риска применения подразделяются на четыре класса. Классы имеют обозначения:
- класс 1 — медицинские изделия с низкой степенью риска;
- класс 2а — медицинские изделия со средней степенью риска;
- класс 2б — медицинские изделия с повышенной степенью риска;
- класс 3 — медицинские изделия с высокой степенью риска.
Классификация медицинских изделий для диагностики in vitro:
- класс 1 — медицинские изделия с низким индивидуальным риском и низким риском для общественного здоровья;
- класс 2а — медицинские изделия с умеренным индивидуальным риском и/или низким риском для общественного здоровья;
- класс 2б — медицинские изделия с высоким индивидуальным риском и/или умеренным риском для общественного здоровья;
- класс 3 — медицинские изделия с высоким индивидуальным риском и/или высоким риском для общественного здоровья.
Размер государственной пошлины
В соответствии с классом определяется размер государственной пошлины за проведение экспертизы качества, эффективности и безопасности медицинских изделий.
Государственная регистрация медицинских изделий
Чтобы зарегистрировать и сертифицировать медицинское изделие, необходимо пройти сквозь сложную вереницу процедур. Государство требует ответственного подхода к здоровью граждан, поэтому создало многоуровневую систему проверок.
Для того, чтобы их пройти, нужны не только знания, но и опыт в производстве и государственной регистрации медицинских изделий.
Нормативно-правовое основание регистрации
Закон «Об основах охраны здоровья граждан» в РФ гласит, что реализовывать медицинские продукты можно лишь при наличии регистрационного удостоверения. Только по окончанию процедуры регистрации медицинское изделие можно признать безопасным и действенным.
Государственное регистрационное удостоверение является специальным документом, который подтверждает, что ваше изделие качественный и безопасный, что его можно использовать в медицинских целях. Без него государство запрещает совершать импорт и экспорт, продавать, производить и использовать средство на всей территории России. Не имея в арсенале данного документа, вы рискуете понести наказание перед законом.
Оформлением удостоверений для медицинских изделий на территории России занимается Росздравнадзор. Там можно получить регистрационное досье, которое необходимо направить в «Национальный институт качества». Или же во «Всероссийский научно-исследовательский и испытательный институт медицинской техники». Там определяется соответствие продукта всем необходимым требованиям.
Постановление №1416 «Об утверждении Правил регистрации медицинских изделий» определяет алгоритмы для выдачи удостоверения для медицинского товара.
Но законодательство о здравоохранении периодически меняется, в постановление постоянно вносятся новые поправки.»Sigma Lab» наблюдает за поправками в постановлениях и законах, чтобы составлять исключительно актуальные документы, которые исключают неприятные контакты с государственными организациями. Мы всегда знаем, что происходит в области здравоохранения, и как составить документ, который государство примет без поправок.
Сроки регистрации
Медицинские изделия классифицируются на 4 группы по степени риска для здоровья:
- Класс 1 – низкая степень риска и опасности;
- Класс 2а – средняя степень риска и опасности;
- 2б – повышенная степень риска и опасности;
- Класс 3 – высокая степень риска и опасности.
Наши специалисты помогут вам точно определить, к какому классу относится ваш продукт. Также эксперты составят список документов конкретно для вашего медицинского продукта; для класса опасности, к которому он принадлежит.
Класс опасности в медицинских изделиях влияет на продолжительность регистрации. Чем выше класс риска и опасности, тем больше времени займет процесс, ведь высокие классы требуют особого внимания.
Специалисты из «Sigma Lab» разрабатывают пакет документов преимущественно для 3-го класса, для изделий с самым высоким уровнем риска. Например, для лекарственных инъекционных средств в косметологии, имплантатов, противоспаечных гелей и других потенциально опасных медицинских изделий.
Наша компания разрабатывает пакеты документов мед изделий 3го класса риска, к которому относятся такие продукты, как имплантаты, косметологические препараты для инъекций, противоспаечные гели и т.п.
Документы, необходимые для регистрации медицинского продукта
|
Изделия, произведенные в РФ |
Изделия, произведенные в других странах |
|---|---|
|
ИНН, ОГРН, выписка из ЕГРЮЛ |
Документ, который подтверждает регистрацию производителя как юридического лица в стране, резидентом которой он является |
|
Сертификат соответствия СМК |
Сертификат изготовителя |
|
Полное описание вашего продукта |
Разрешение |
|
Информация об организации, которая выпустила товар |
Доверенность на представителя организации-производителя в РФ |
|
Инструкция |
Документы с условиями изготовления медицинских изделий |
|
Итоги проведения испытаний |
Инструкция по применению МИ |
|
Фото товара |
Фото |
Список является предварительным, ориентировочным, так как разные проекты имеют разную сложность реализации. Эта сложность определяется классом опасности и риска, а также функциональными особенностями медицинских средств.
Обратитесь за консультацией к нашим экспертам, чтобы узнать пакет документов, который потребуется для регистрации медизделия конкретно в вашем случае.
Как проходит разработка документации в ООО «Sigma Lab»?
Первый этап. Составление технической документации
- Файл технических условий для регистрации медицинских изделий;
- Технический регламент производства;
- СОП (стандартная операционная процедура), соответственно стандартам ГОСТ ISO 13485-2017;
- Перечень методик и проведение испытаний, акт квалификационных испытаний.
Второй этап. Составление регулирующей документации
- Файл менеджмента риска на продукт;
- Инструкция по применению продукта;
- Перечень нормативной документации.
Третий этап. Составление сопроводительной документации
- Фотоальбом (ваши изделия с маркировкой, комплектация, все варианты исполнения, производство);
- Заявление, пояснительная записка, комплект документов с копиями.
Сотрудничество с ООО «Sigma Lab»
Мы оказываем помощь при регистрации изделий медицинского назначения. В наши услуги входит:
- Проверка и внесение коррективов в предоставленные бумаги;
- Организация процесса регистрации на любом его этапе;
- Консультации во время испытаний продукции.
При необходимости мы обеспечиваем полное сопровождение при регистрации изделия, также возможны и разовые консультации.
При заказе комплексного обслуживания вы можете не беспокоиться о процессе. Вам будет необходимо только предоставлять необходимые документы, остальное же мы возьмем на себя.
Все отношения между компанией «Sigma Lab» и заказчиком регулируются официальным договором. Благодаря этому вы можете быть уверены в качестве услуг и быть в курсе того, что конкретно мы обязуемся сделать для регистрации изделия.
Разовые услуги подойдут вам в том случае, если вашей компании необходима вводная консультация, проверка при получении удостоверения и т.п.
Плюсы сотрудничества с компанией «Sigma Lab»
Мы ценим время клиентов, поэтому сокращаем время регистрации до минимума. Пока мы ведем ваши дела, вы можете заниматься другими, не менее важными вопросами для вашего бизнеса. Наши специалисты же сделают все, чтобы вы смогли как можно быстрее вывести свой товар на рынок.
Типичные консалтеры работают только с документами. Мы же участвуем и в производстве медицинских продуктов. Этот опыт позволяет нам работать над регистрацией без вашего участия. С вашей стороны будет необходимо определить состав продукта, фасовку и дизайн.
Чтобы получить качественную консультацию или комплексное сопровождение при регистрации медицинского изделия, заполните форму ниже. Мы будем рады ответить на любой ваш вопрос!
Кроме того, компания Sigma Lab является научно-производственным предприятием, в задачи которого входит разработка, прототипирование и запуск серийного производства медицинских изделий в жидкой форме на основе биополимеров. Мы всем необходимым оснащены качественным современным оборудованием для выполнения подготовки, розлива, укупорки, упаковки и стерилизации медицинской продукции в соответствии со стандартами GMP и ISO 13485.
Регистрационный номер — кодовое обозначение, присвоенное лекарственному препарату при его государственной регистрации. (ФЗ № 61 от 12.04.2010 г)
Регистрационное удостоверение лекарственного препарата — документ, подтверждающий факт государственной регистрации лекарственного препарата. (ФЗ № 61 от 12.04.2010 г)
При первой регистрации препарата в России регистрационное удостоверение выдается на 5 лет.
По истечении данного срока производитель подает документы для подтверждения регистрации препарата и тогда уже регистрационное удостоверение выдается бессрочно.
Держатель или владелец регистрационного удостоверения лекарственного препарата — разработчик лекарственного средства, производитель лекарственных средств или иное юридическое лицо, обладающее правом владения регистрационным удостоверением, которые несут ответственность за качество, эффективность и безопасность лекарственного препарата.
(ФЗ № 61 от 12.04.2010 г)
СТАНДАРТИЗАЦИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ
В соответствии с ФЗ № 61 от 12.04.2010 г.:
Качество лекарственного средства — соответствие лекарственного средства требованиям фармакопейной статьи либо в случае ее отсутствия нормативной документации или нормативного документа.
Безопасность лекарственного средства — характеристика лекарственного средства, основанная на сравнительном анализе его эффективности и риска причинения вреда здоровью.
Эффективность лекарственного препарата — характеристика степени положительного влияния лекарственного препарата на течение, продолжительность заболевания или его предотвращение, реабилитацию, на сохранение, предотвращение или прерывание беременности.
Стандартизация ЛС — разработка и применение унифицированных требований и методов исследования лекарственных форм (стандартов).
Стандарт качества ЛС — нормативный документ, содержащий перечень нормируемых показателей и методов контроля качества лекарственных средств, утверждаемый Министерством здравоохранения Российской Федерации (Минздравом России).
Стандарты качества ЛС подразделяются на две категории:
1) Государственные стандарты качества ЛС — Общая фармакопейная статья (ОФС) и Фармакопейная статья (ФС);
Фармакопейная статья предприятия (ФСП).
Общая фармакопейная статья — документ, утвержденный уполномоченным федеральным органом исполнительной власти и содержащий перечень показателей качества и (или) методов контроля качества конкретной лекарственной формы, лекарственного растительного сырья, описания биологических, биохимических, микробиологических, физико-химических, физических, химических и других методов анализа лекарственного средства.
Фармакопейная статья — документ, утвержденный уполномоченным федеральным органом исполнительной власти и содержащий перечень показателей качества и методов контроля качества лекарственного средства.
Фармакопейная статья предприятия — это стандарт качества
лекарственного средства на лекарственное средство под торговым названием, содержащий перечень показателей и методов контроля качества лекарственного средства производства конкретного предприятия, учитывающий конкретную технологию данного предприятия и прошедший экспертизу и регистрацию в установленном порядке.
Государственная фармакопея (ГФ) — это сборник государственных стандартов качества ЛС, имеющий законодательный характер.
СЕРТИФИКАЦИЯ И ДЕКЛАРИРОВАНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ И МЕДИЦИНСКИХ ИЗДЕЛИЙ.
С целью подтверждения соответствия товара определенному уровню качества, указанному в НТД, проводится сертификация и декларирование соответствия.
Объекты обязательной сертификации определяются Постановлением Правительства РФ № 982 от 1 декабря 2009 г.(ред. 18.06.2012 № 596) «Об утверждении единого перечня продукции, подлежащей обязательной сертификации, и единого перечня продукции, подтверждение соответствия которой осуществляется в форме принятия декларации о соответствии».
Обязательное подтверждение соответствия качества товара осуществляется в двух формах — путем обязательной сертификации, путем принятия декларации о соответствии.
ЛС относятся к видам продукции, несущим потенциальную опасность. Это объясняется тем, что некачественные или фальсифицированные лекарства могут принести вред здоровью.
В единый перечень продукции, подлежащий обязательной сертификации включенынекоторые виды товаров класса 9300 «Медикаменты, химико-фармацевтическая продукция и продукция медицинского назначения» Общероссийского классификатора продукции (ОКП):
— Сыворотки, иммуно- и гаммаглобулины, препараты из крови прочие и полученные методом генетической инженерии, применяемые в медицине;
— Вакцины, анатоксины и токсины, применяемые в медицине.
— Бактериофаги.
В единый перечень продукции, подтверждение соответствия которой осуществляется в форме принятия декларации о соответствии включены:
— Лекарственные средства, зарегистрированные в установленном порядке, (кроме указанных в предыдущем перечне);
— Оборудование медицинское;
Как проверить легальность и регистрацию лекарства, БАДа?
Каждый день появляются все новые лекарственные препараты их рекламируют по телевидению и в интернете, но не каждое, рекламируемое лекарство несет в себе ту пользу, которую так красочно расписывают в рекламе.
Как не купить пустышку и не легальный лекарственный препарат?
Каждое разрешенное в России лекарство, должно иметь государственную регистрацию Минздраве России и быть в реестре лекарственных средств. Зачем нужна регистрация лекарственных препаратов? Регистрируя препарат, государство берет на себя ответственность за проверку его эффективности и безопасности, плюс проводятся локальные клинические исследования, даже если они уже проводились в других странах.
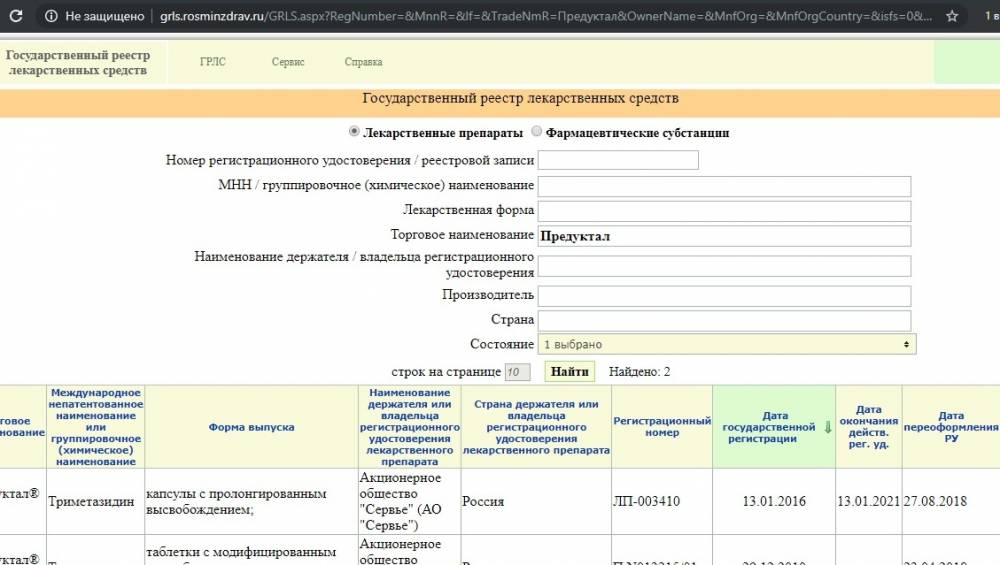
Как проверить лекарство через Минздрав России?
Государственный реестр лекарственных средств
Вводите торговое наименование и нажимаете найти. Если препарат есть реестре, то внизу вы увидите таблицу с данными всех производителей, форм выпуска, даты регистрации и т.п.
Как проверить лекарство на сайте Росздравнадзора?
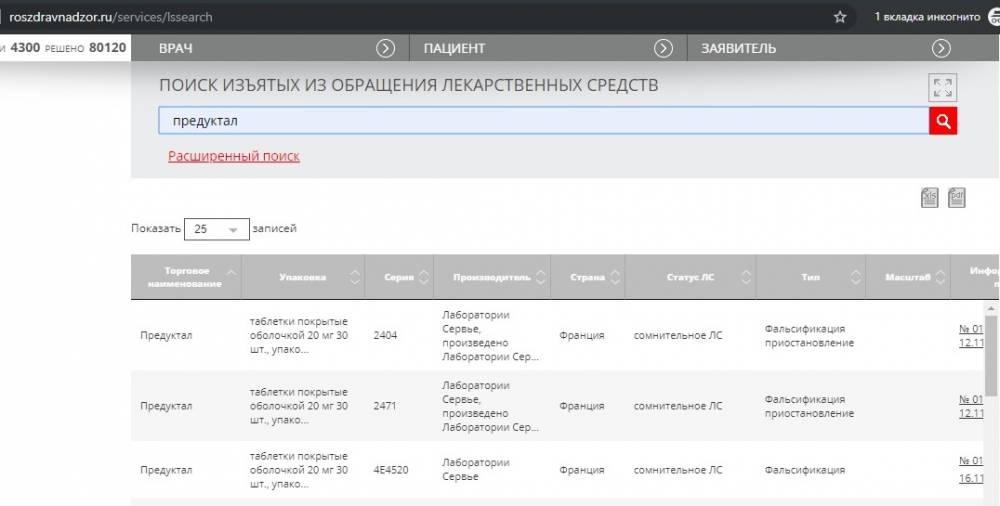
Используя поиск изъятых из обращения лекарственных средств, можно проверить лекарство на подлинность, на подделку. Для точного результата, нужно использовать «расширенный поиск» указывая серию, дату.
Как проверить БАДы на подлинность на сайте Роспотребнадзора?
БАД относится к компетенции Роспотребнадзора. Получить сведения о биологически активных добавках, прошедших государственную регистрацию и разрешенных к ввозу и обороту на территории Российской Федерации, можно на официальном сайте Роспотребнадзора.
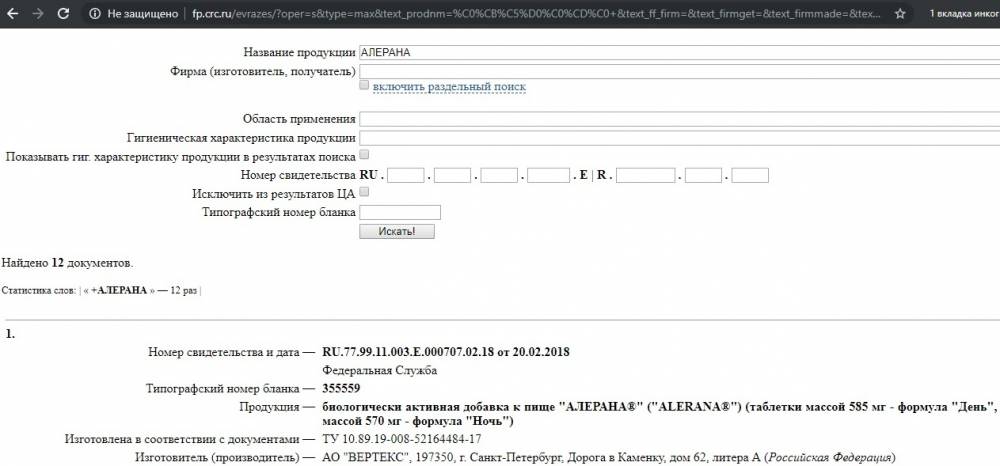
Реестр свидетельств о государственной регистрации (единая форма Таможенного союза, российская часть).
Также там можно проверить и парфюмерно-косметическую продукцию и продукты питания и бытовую химию и многое другое. Если нажать «полный список» вы увидите весь перечень, проверяемых наименований.
Как проверить лекарство и БАД через справочник лекарств РЛС?
РЛС® — Регистр лекарственных средств России. Энциклопедия лекарств и товаров аптечного ассортимента
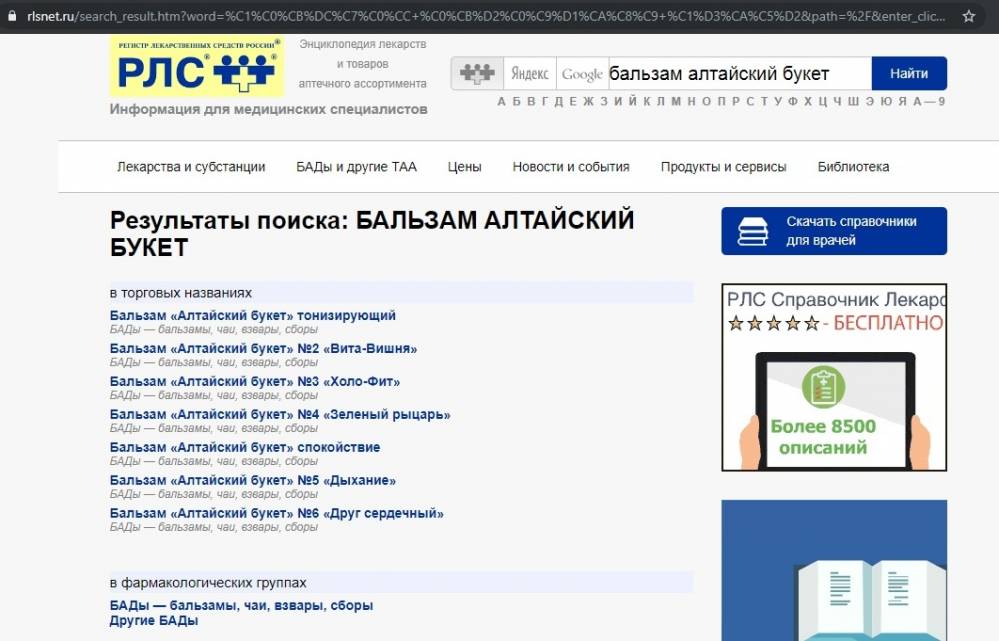
Здесь можно узнать является препарат лекарством или бадом, действующие вещества, наличие в аптеках. Причем если вы укажете в поиске действующее вещество, то получите полный список аналогов препаратов с данным веществом.
***
В нашем стремительно развивающемся мире и засилье информационных технологий, нужно быть на чеку. Не доверять красивой рекламе и красочной упаковке. Нужно искать информацию, отзывы, проверять все самому. Только так можно добиться безопасности и не попасться на удочку производителей контрафакта.
Правительство утвердило перечень медицинских изделий для упрощенной госрегистрации
Перечень включает 36 видов медицинских изделий: костюмы изолирующие и хирургические, халаты изолирующие и операционные, наборы одежды хирургический и смотровой, маски, бахилы, респираторы, перчатки.
Документ позволит в короткие сроки вводить в обращение медицинские изделия, необходимые для предупреждения распространения инфекционных заболеваний, говорится в пояснении к постановлению.
Сегодня на заседании правительства премьер-министр Михаил Мишустин подчеркнул, что российская промышленность должна увеличить объемы производства и ускорить выпуск медицинских изделий, на которые сейчас есть повышенный спрос.
«Минздраву необходимо определить порядок их упрощенной, а значит, более быстрой регистрации. Минпромторгу необходимо передать в правительство график, как мы договорились, ежедневных объемов производства масок и других средств индивидуальной защиты с учетом ввода новых мощностей. Мы будем в режиме онлайн контролировать ситуацию с их наличием. И для этого создаем единый информационный центр, где будут собираться все сведения по каждому региону», – оповестил премьер.